
02/04/2021
VACCINI E RISCHIO TROMBOSI
I vaccini nella loro generalità, hanno il fine di stimolare il [...]
Il nostro sistema immune ha sviluppato la capacità di proteggerci da una pluralità di agenti infettivi mettendo in atto un meccanismo di difesa innato in grado di avvertire già i primi segni di una infezione e di innescare un sistema adottivo, sia cellulare che umorale, nel tentativo di eradicare l’agente infettante.
A seguito di una prima infezione talora il nostro organismo sviluppa una memoria che ci protegge da ulteriori attacchi dallo stesso virus o da certe sue varianti.
Purtroppo non tutti i virus conferiscono una immunità protettiva ma ci lasciano soggetti ad ulteriori reinfezioni dello stesso virus. Il meccanismo di difesa messo in atto, nonostante riesca molto spesso ad eradicare l’infezione, talvolta fallisce a causa di meccanismi che il virus riesce ad evolvere e che gli permettono di aggirare le nostre difese.
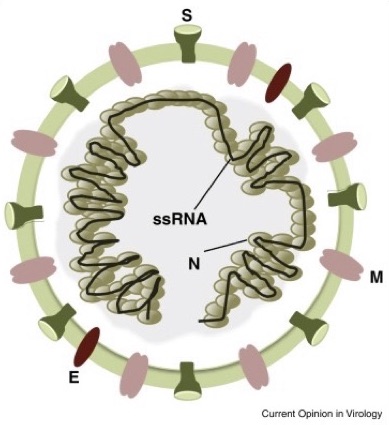
Spike (S), envelope (E), matrix (M) and nucleocapsid (N). Interspersed between these structural proteins are accessory
Per penetrare nelle cellule i Corona virus utilizzano alcune sostanze sulle pareti delle cellule ospiti, diverse secondo il virus presente; i virus della SARS e del COV-19 legano la ACE-2 (carbossi-peptidase) e alcuni acidi salici presenti su gangliosidi della membrana cellulare con la partecipazione anche di proteoglicani, mentre la MERS lega una dipeptidil-peptidasi (DPP4).
Per consentire il legame al rispettivo recettore, la proteina S subisce una rottura proteolitica, da parte di proteasi a serina (TMPRSS2 e HAT), con formazione di una subunità (S2) che consente la fusione del virus alla cellula e la liberazione del RNA nel citoplasma dove il virus viene trasportato in vescicole attraverso il sistema endosomiale/lisosomiale.
Qui le glicoproteine virali sono idrolizzate da proteasi in ambiente acido, per poi andare incontro ad una serie, non ben conosciuta, di trascrizioni, replicazioni e formazione di un nuovo RNA incapsulato in un nucleocapside che viene inglobato in una vescicola trasportata alla superfice e liberata fuori della cellula. La stessa TMPRSS2 opera anche una scissione di alcuni aminoacidi della ACE2 per facilitare la penetrazione nelle cellule.
E’ interessante osservare il legame che il virus ingaggia con il recettore cellulare ACE2, particolare che ha gettato il sospetto che alcuni farmaci antiipertensivi (inibitori dello ACE e del recettore per la Ang II) potessero incrementare il rischio di infezione come conseguenza della possibilità che questi farmaci hanno di indurre la espressione di ACE2.

(La figura riporta la via metabolica dell’angiotensinogeno. Patel VB et al 2016) vir:iMMun copia (trascinato)
Il sospetto che l’aumentata presenza di ACE2 possa rappresentare un fattore di rischio viene tuttavia bilanciato dal fatto che lo ACE2, tramite la produzione di Ang(1-7) esercita un effetto protettivo a livello cardio-vascolare e su altri organi. L’ACE2 è una metalloproteasi contenente un atomo di zinco e quindi attaccabile con chelanti di ioni bivalenti.
L’ingresso del virus è anche favorito da una proteasi a serina (TMPRSS2) endosomiale.
Nonostante i ragionevoli sospetti alcuni dati clinici non rivelano alcun maggiore rischio nei pazienti affetti da COVID19 e trattati con gli antiipertensivi ARBs/ACEi, oltre al fatto che questi ultimi pazienti hanno presentato una ridotta risposta infiammatoria e una più mite progressione della malattia.
Vale la pena ricordare come la Ang(1-7), indotta da ACE2, abbia una attività antinfiammatoria e inibitoria sulla espressione di IL-6.
Vi sono infine alcuni dati sperimentali dai quali risulterebbe che una maggiore presenza in circolo di ACE2 potrebbe fungere da decoy per le cellule virali inibendone la funzione.
Una volta che la cellula è stata infettata varie molecole presenti sul virus (PAMPs) vengono riconosciute da diversi recettori cellulari (TLRs, PRRs, RLRs ed altri) che danno inizio ad una rapida risposta immunitaria.
Oltre alla risposta delle cellule dell’epitelio respiratorio anche le cellule dendritiche (DCs) intraepiteliali e i macrofagi ampliano la risposta.
A questa prima fase segue una risposta delle cellule T, iniziata dal riconoscimento da parte delle cellule dendritiche respiratorie degli antigeni virali, con migrazione ai linfonodi per indurre proliferazione clonale di specifiche cellule T e loro migrazione al sito infetto. E’ a questo punto che si amplia la produzione di citokine, chemokine, PGD2 oltre a molecole citotossiche che possono, direttamente o indirettamente, inibire la replicazione virale.
Nei casi di infezione più avanzata si può verificare leucopenia e linfopenia,
alterata attivazione delle cellule T, scarsa risposta anticorpale.
Gli attacchi da Corona virus sono generalmente caratterizzati da una prolungata espressione di citokine che contribuiscono a generare una tempesta lesiva per organi e tessuti.
La scarsa risposta anticorpale fa nascere il sospetto che siano possibili anche reinfezioni da parte dello stesso virus anche dopo tempi brevi.
La risposta degli organismi infettati può essere diversa e dipende dal tipo di virus e dalle strategie che questi mettono in atto per inibire la risposta immune innata che, usualmente, prevede la produzione di IFN I e con il riconoscimento dei recettori per IFN a/b a generare un ambiente ostile al virus, unitamente alle cellule NK.
Tuttavia, nel caso di infezione da COV-19 si riscontra una scarsa espressione di IFN tipo I con le conseguenze che questa determina. I macrofagi attivati tendono a fagocitare le cellule infette e i materiali derivanti dalla apoptosi delle stesse cellule per prevenire un una ulteriore produzione di chemokine e il reclutamento di altre cellule infiammatorie. Tuttavia in presenza di una eccessiva infezione i macrofagi possono perdere la loro efficacia con conseguente accumulo di cellule infettate, cellule infiammatorie, proteine del siero e residui apoptotici che possono condurre ad una grave bronchiolite.
In maniera analoga le DCs, unitamente ai complessi antigene-MHC, migrano ai linfonodi attraverso i vasi linfatici secondari dove interagiscono con i linfociti CD4+ e CD8+ che proliferano e differenziano in cellule T effettrici dirette verso i tessuti infetti.
La imponente risposta infiammatoria è il primo evento nocivo indotto dal virus che, una volta penetrato in circolo ingaggia un altro drammatico attacco alla struttura della emoglobina tramite alcune proteine e glicoproteine non strutturali presenti sulla membrana del virus:(orflab, ORF3a, ORF6, ORF8, ORF10).
Grazie a queste specifiche proteine il virus, ormai dentro l’eritrocita, attacca il gruppo EME sulla catena beta della emoglobina, lega la porfirina e dissocia il ferro lasciando una emoglobina che non può più provvedere agli scambi O2/CO2.
Le cellule polmonari subiranno una intossicazione e una forte infiammazione che determina un alterato aspetto del polmone.

(fonte immagine da Internet-vir:iMMun copia (trascinato) 2)
Questo evento va anche ad interferire sul normale metabolismo del gruppo eme mentre la porfirina legata alle proteine del virus non subirà la sua naturale metabolizzazione epatica in biliverdina e bilirubina, che rappresenta uno dei più efficienti antiossidanti del nostro organismo.
Nel contempo vi sarà un accumulo di ioni ferro che causeranno ancora infiammazione e ulteriori danni ossidativi, mentre aumenterà la presenza di ferritina nel tentativo di ridurre il danno degli ioni ferro liberi.
Questo attacco ha come conseguenza una presenza di emoglobina in circolo, priva di ferro, e pertanto incapace di veicolare ossigeno e anidride carbonica, incapacità che determina una forte ipossiemia oltre a maggiore tossicità e infiammazione.
In questa condizione l’organismo cerca di compensare la carenza di ossigeno mettendo in circolo la eritropoietina per aumentare la presenza di globuli rossi.
In effetti viene riscontrato un aumento di emoglobina ma con bassa saturazione di ossigeno e quindi inefficiente.
Il quadro è complicato, come già detto, da un sistema immune alterato e da una imponente reazione infiammatoria responsabile di numerosi danni d’organo.
I polmoni diventano incapaci di ostacolare lo stress ossidativo e la carenza di ossigeno, mentre anche il fegato è oberato nel tentativo di eliminare il ferro e libera alanina amino-trasferasi.
Fortemente coinvolto risulta il cuore sia per la presenza della ACE2 sulle sue cellule, sia per i danni legati al forte processo infiammatorio e ai processi trombolitici che vengono riscontrati.
Viste le scarse conoscenze sul COVID-19, per ora disponibili, dobbiamo considerare quanto sopra riportato come una possibile, ma realistica, ipotesi.
Tuttavia se consideriamo questi eventi come reali, possiamo comprendere anche i potenziali interventi per neutralizzare la patogenicità del virus o almeno per ridurne la gravità.
Ovviamente la realizzazione di uno specifico agente antivirale o un efficace vaccino sono gli obiettivi primari, tuttavia, in attesa di questi, vi sono alcune fasi della infezione potenzialmente sensibili anche per farmaci già conosciuti.
Un primo intervento potrebbe essere contrastare le proteine che consentono la penetrazione del virus nelle cellule: una competizione per la ACE2 di membrana o alla proteina-a-serina TMPRSS2 che partecipa all’ingresso nelle cellule.
Ridurre in tutti i modi lo stress citokinico e ossidativo facendo ricorso ai vari farmaci già disponibili e idonei a modulare la abnorme reazione infiammatoria.
Impiegare alcuni farmaci antivirali utilizzati per altre infezioni (Favipiravir, Remdesivir…) ma che potrebbero avere un effetto, sia pure ridotto, anche verso il COV-19.
Infine ricorrere alla Idrossiclorochina (HCL) sfruttando alcuni dei meccanismi di azione che questo farmaco mette in atto anche nei confronti del plasmodio della malaria. In effetti la HCL si lega con alta affinità agli acidi sialici e ad alcuni gangliosidi di membrana limitando il legame della proteina virale Spike con i recettori cellulari. La HCL, grazie ad una sua azione alcalinizzante, inibisce anche il transito endosomiale e nelle vescicole del Golgi del virus limitandone la replicazione. Alcune analogie con il meccanismo messo in atto nei confronti del plasmodio della malaria, hanno portato ad evidenziare come la HCL impedisca anche il legame di alcune proteine virali al gruppo porfirinico della emoglobina e al distacco del ferro dalla porfirina stessa. Questo meccanismo viene messo in atto dal virus per indurre lo stato ipossemico tipico della patologia da COV-19.
La Idrossiclorochina ha anche una ben nota azione anti-infiammatoria e immunomodulante che si traduce in una inibizione della espressione di varie citokine incluso il TNF-alfa e la IL-6 considerata di particolare significato patogenetico.
Possono inoltre essere di ausilio, alcuni antibiotici che assommano anche proprietà anti-infiammatorie (Azitromicina, Claritromicina), ma anche certi
inibitori della attivazione del fattore NFkB, gli inibitori delle kinasi JAK1/2, gli anti-ossidanti classici (Vitamina C, glicirrizina , zinco…) e trasfusioni di sangue con anticorpi anti-proteina S, ma in particolare, dosi adeguate di eparina per prevenire le frequenti coagulopatie e per una possibile interferenza sulla adesione del virus alle pareti cellulari per un effetto competitivo con alcuni proteoglicani di superficie.
Infine la Vitamina D3 per i suoi effetti modulatori del sistema immune (inibizione di varie citokine e PGE2), liberazione di defensine e catelecidine.
Vi sono anche altre potenziali opzioni che vengono periodicamente riportate dalla letteratura e in fase di studio.
Ovviamente, al momento, si può parlare solo di ipotesi terapeutiche mentre per molti di questi farmaci sono in corso studi clinici atti a valutare la potenziale applicazione in questa gravosa infezione, magari con effetti adiuvanti o semplicemente per ridurre la gravità della patologia, in attesa di un idoneo vaccino o di un efficiente farmaco antivirale.
Livorno, 28-03- 2020 (S.Rosini )
I vaccini nella loro generalità, hanno il fine di stimolare il [...]
EVIDENZA DI INFETTIVITA’ DEL CAVO ORALE Nonostante sia ben conosciuto come [...]